Methods for Describing Column Efficiency
There are two related terms used as quantitative measures of chromatographic efficiency:
- Number of ‘theoretical plates’ N
- Plate height H.
These parameters are used to measure a column’s resolving power (ability to differentiate between closely migrating peaks - sometimes called efficiency). The two parameters are related by the following equation:
where L is the length of the column packing (usually in cm - this is a constant for a given column).
The term theoretical plates refers to an early explanation of the resolving power of a column where the column was viewed as a ‘stack’ of closely related but discrete layers (or plates). The more plates in the column the greater the resolving power of the column but as the column has a definite length, more plates equals thinner plates. In other words N and H are inversely proportional. As N increases (for a given column) H decreases.
There are great differences in the plate height and numbers of theoretical plates between columns due to differences in the column type as well as the stationary and mobile phases used.
Thus variations of the following magnitudes exist:
| Plate height: | 0.1 to 0.001 mm or smaller |
| Plate numbers: | 100 to 100 000 |
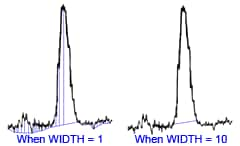
There are a number of complex methods for determining N but a simple approximation of N is given by:
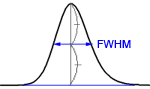
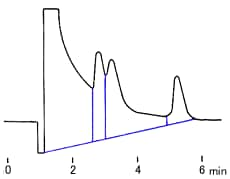
where W/2 = half of the width of the peak at its base, which is also equal to the width of the peak at half its maximum height and tg = retention time (time taken for the peak to come off the column) of the analyte.

Sample problem:
Calculate the number of theoretical plates N and the plate height H, when the retention time is 20.40 minutes, half of the base width (given in minutes) is 0.65 minutes and the column length is 30 cm.
Solution
Practical application of N
N is usually used to measure and verify the performance of a column.
Measurement of N can be used to:
- measure the resolving power of a column
- measure that the resolving power of a new column is to specification
- monitor the resolving power of a column over time
- determine if column replacement is required due to deterioration in performance
- troubleshoot problems.
The number of theoretical plates
Gas chromatography columns normally have
The number of theoretical plates is related to the retention time,
If the peaks are reasonably symmetric, it can be assumed that they have a Gaussian shape. Then
where
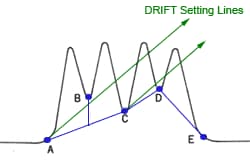
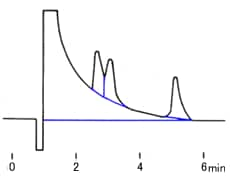
You find the peak width at half height by drawing a line vertically from the peak maximum to the baseline, measuring half-way up the peak, drawing a horizontal line, and measuring the length of the horizontal line.
You measure the retention time (designated avove as
Both
other hand-----
EXAMPLE
A chromatogram from a certain column has a peak with a
Solution
Analytical Wavelength |
The analytical wavelength for quantitative analysis is normally max ie the wavelength corresponding to an absorption peak.
 |
There are several reasons for this choice.
Absorbance per unit concentration of analyte is greatest at max - this provides maximum sensitivity. That is to say, the higher the absorbance, the lower the concentration that the analysis is able to reliably measure.
Consider the comparison (below) of using the analytical wavelength at max and at half
max - the analyte concentration is some value called ‘x’. If absorbances cannot be read below 0.1 because of the potential for substantial error, you can see in this example that
max enables the analyte concentration to be determined down to at least x/8 whereas this concentration falls below the 0.1 absorbance threshold at half
max. The higher the absorbance per unit of concentration (highest at
max), the more sensitive the analysis.

The absorbance curve is normally flat (or plateaus out) around max and this circumstance leads to optimum precision and accuracy. There can be some spectrophotometer error in the setting of
max but without there being significant change in the value of the absorbance. On the other hand, if the analytical wavelength is selected from a point on the side of a peak, the same error in wavelength setting can have a significant impact on absorbance. Error in the wavelength setting of a spectrophotometer translates to imprecision/inaccuracy in the calculated concentrations.